
Christopher R. Iacovella
About Me
For the past two decades, I’ve been immersed in the field of molecular modeling and simulation, performing research on a wide range of topics. In the last few years, my research has focused primarily on three main areas (1) rational design of thin-films, (2) multiscale modeling of skin lipid membranes, and (3) the development of the Molecular Simulation and Design Framework (MoSDeF) for the reprodicible initialization of complex systems.
Biographical sketch:
- Bioinformatics Software Engineer (2023-current)
- Memorial Sloan Kettering Cancer Center
- Chodera Lab
- AI-driven Structure-enabled Antiviral Platform (ASAP)
- Research Assistant Professor (2012-2023)
- Vanderbilt University
- Department of Chemical and Biomolecular Engineering
- Center for Multiscale Modeling and Simulation
- Postdoctoral Research Associate (2009-2012) — Vanderbilt University
- Department of Chemical and Biomolecular Engineering
- Postdoctoral advisor: Peter T. Cummings
- Visiting Junior Scientist (2011)
- National Academy of Sciences of Ukraine
- Institute for Condensed Matter Physics
- Ph.D. Chemical Engineering (2003-2009)
- The University of Michigan
- Graduate thesis advisor: Sharon C. Glotzer
- B.S. Chemical Engineering (1999-2003)
- The University at Buffalo
- Undergraduate research advisor: David Kofke
Research
Over the years I’ve been involved in a wide range of topics (see Publications), including: Development of software to enable large-scale screening and reproducible simulations, nanoscale lubrication via thin films, application of machine learning to nanoscale lubrication, multiscale modeling of skin lipid membranes, design of solvent-adaptable films, atomistic and coarse-grained force field development, nanoparticle-cell membrane permeation, phase behavior of self-assembled nanoparticles, lipid self-assembly, model development for carbide-derived carbons, multiscale modeling of molecular electronic junctions, failure mechanisms in nanowires, free energy calculations of nanoconfined fluids, microfluidic packing of colloids, colloidal interactions, and development of order parameters. Summaries of my more recent research directions are below.
Molecular Simulation and Design Framework (MoSDeF)
Enabling reproducible science is fundamentally important. While the simulation community has largely shifted to using community developed simulation engines-—a huge leap in terms of ensuring reproducibility—-initialization and parameterization of systems has still largely been performed using in-house software/scripts that are not freely available. Without access to this code, important initialization procedures/choice may be obscured making reproduction of a given simulation not possible. Without viewing the source, the quality of the code is unknown (does it contain sufficient testing?) and any bugs/errors will remain undiscovered. Furthermore, classical simulations that rely on force fields are particularly challenging for reproducibility. Simply citing a paper as the source of the parameters, as is common practice, is often insufficient as it does not provide information about which specific parameters were used and under what circumstances (i.e., it does not convey the logic used for applying the parameters), nor does it indicate conversion factors. Given the complexity of force fields, different users may interpret the force field differently and may ultimately utilize different parameters. Errors related to human error may also go unnoticed, such as typographic or unit conversion errors; these often will not result in failure of a simulation, but rather implementation of a system that does not faithfully reproduce the original model. Furthermore, creating these initialization and parameterization codes can often be very time consuming, preventing scientists from focusing on exploring the larger scientific questions of their research.
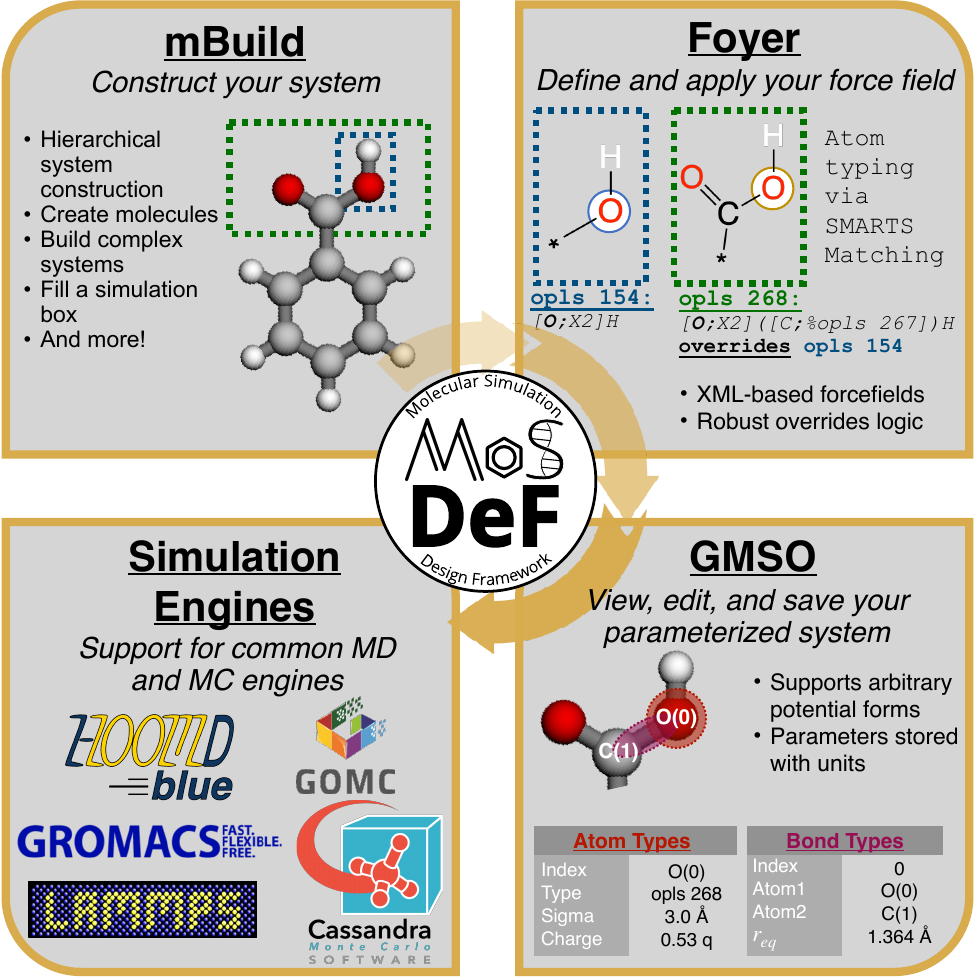
MoSDeF was created to address many of these issues, providing a set of extensible Python libraries for the construction, parameterization, and generation of syntactically correct input files for simulation engines. MoSDeF includes 3 key components:
- the mBuild library for the hierarchical construction of system configurations
- the foyer library for encoding and applying force field parameters and
- the GMSO library for storing parameterized systems in a simulation engine agnostic format, with translators to write data files for various simulation engines.
mBuild is unique in that it allows systems to be constructed from smaller, interchangeable pieces (“Compounds”), rather than using a rigid data-structure hierarchy. It provides routines to rotate, translate, scale, and duplicate Compounds. mBuild supports defining bonds explicitly or defining “Ports”, allowing separate Compounds to be joined later (for example, allowing arbitrary polymerization by encoding the procedure in a loop); mBuild include routines to automatically orient Compounds when connecting Ports to simplify the construction of complex systems.
Foyer provides a scheme for defining both parameters and their usage rules in a single XML file. Usage rules are defined via SMARTS strings (that define the chemical environment for which a parameter applies) and overrides statements to define rule precedence. In foyer, all rules are evaluated, adding atom-types whose rules match the given atom to a white list, and those that do not match (or appear in an override statement for a matching atom-type) to the black list; for a sufficiently defined force field, the intersection between white and black lists will yield a single applicable atom-type. This scheme provides several advantages:
- Usage rules are both human and machine readable
- the use override statements means that the order rules appear in a file does not matter
- all rules in the file are evaluated for each atom (i.e., there is no hierarchy), helping to ensure that force field rules are fully defined and unambigous
- by placing rules in an XML file, rather than within the source code, force fields can be easily shared, extended, evolved, and version controlled,
- changes to the force field (or creation of custom force fields) does not require changes to the underlying engine used to evaluate them.
GMSO is used to store the parameterized structure in an engine agnostic format that stores all the relevant positions, bonds, angles, etc. and the associated parameters. Unique to GMSO is the use of symbolic math (SymPy) to define the functional form of each interaction, allowing it to store parameters with effectively any functional form. When writing a data file for a specific engine, GMSO automatically checks to ensure that the provided functional forms are supported by the engine. The use of symbolic math also allows otherwise identical functions that are simply encoded in different forms to be identified. Sympy can also identify scaling factor differences (e.g., some simulation codes expected the harmonic potential to have a factor of 1/2, while others lump this into the spring constant) and then automatically scales the give parameters accordingly. Throughout MoSDeF the unyt package is used such that parameters have explicit units associated with them and can automatically and correctly converted to the unit expected by a simulation engine. But saving all parameters in a single format with no data loss, the code relies upon on 1 way conversion (i.e., from GMSO to the target format), rather than translating from one engine format to another; information may be lost upon writing to a target file requiring a translator to assume information, e.g., units, which can give rise to errors.
Since the entire set of the libraries are written in Python, they can easily interface with other python packages (or packages with python bindings). Currently, wrappers are included to various tools already, including RDKit (allowing Compounds to be initialized from SMILES strings), packmol for filling boxes, openBabel for energy minimization, and others. The design allows all steps in a workflow to be scripted, which in itself, aids in reproducibility (i.e., all steps taken in a workflow are automatically captured) and also enables automation, such as screening over a variable in the system such as number of repeat units in a polymer, the chemistry of the repeat unit, system size.
Related links:
- MoSDeF webpage - MoSDeF.org
- Github landing page for MoSDeF - https://github.com/mosdef-hub/
- Cell list written in Python compatible with mbuild Compounds, from my personal github - mbuild_cell_list
- Python library to read legacy hoomdxml file and convert to mbuild Compounds from my personal github - hoomdxml_reader
Related publications
- Cummings PT, McCabe C, Iacovella CR, Ledeczi, A, Jankowski E, Jayaraman A, Palmer JC, Maginn EJ, Glotzer SC, Anderson JA, Siepmann JI, Potoff J, Matsumoto RA, Gilmer JB, Dever RS, Singh R, Crawford B (2021) Open-Source Molecular Modeling Software in Chemical Engineering Focusing on the Molecular Simulation Design Framework, AIChE J. 2021; 67:e17206 DOI:10.1002/aic.17206
- Thompson MW, Gilmer JB, Matsumoto RA, Quach CD, Shamaprasad P, Yang AH, Iacovella CR, McCabe C, Cummings PT (2020) Towards Molecular Simulations that are Transparent, Reproducible, Usable by Others, and Extensible (TRUE), Molecular Physics 118, 9-10, e1742938 DOI:10.1080/00268976.2020.1742938
- Summers AZ, Gilmer JB, Iacovella CR, Cummings PT, McCabe C (2020) MoSDeF, a Python Framework Enabling Large-Scale Computational Screening of Soft Matter: Application to Chemistry-Propery Relationships in Lubricating Monolayer Films, Journal of Chemical Theory and Computation., 16,3,1779-1793 DOI:10.1021/acs.jctc.9b01183
- Klein C, Summers AZ, Thompson MW, Gilmer JB, McCabe C, Cummings PT, Sallai J, Iacovella CR (2019) Formalizing Atom-typing and the Dissemination of Force Fields with Foyer, Computational Materials Science, 167, 215-227 DOI:10.1016/j.commatsci.2019.05.026
- Klein C, Sallai J, Jones TJ, Iacovella CR, McCabe C, Cummings PT (2016) A Hierarchical, Component Based Approach to Screening Properties of Soft Matter, Molecular Modeling and Simulation: Applications and Perspectives, Foundations of Molecular Modeling and Simulation, pp 79-92 DOI:10.1007/978-981-10-1128-3_5
Rational Design of Thin Films
Thin films have been proposed as a means to provide lubrication for nanoscale contacts. Several challenges arise when using thin films as lubricants: (1) films tend to degrade rapidly while undergoing shear and (2) the vast parameter space of possible film chemistries makes identifying films that simultaneously demonstrate low coefficients of friction and low adhesion forces challenging using experiment.
This research has focused on developing a more robust understanding of how model ideality/non-ideality influences the structural and tribological behavior. For example, comparing films attached to crystalline silica substrates (where all molecules are equally spaced) compared to amorphous silica (where spacing may vary). The role of surface roughness was also explored, resulting in the development of a synthesis mimetic procedure using reactive MD to replicate surface etching in experimental resulting from washing substrates with “Piranha solution” (a mixture of sulfuric acid and hydrogen peroxide). The relationship between surface roughness and film breakdown were also explored along with cross-linking.
To examine the vast landscape of film chemistries, the MoSDeF library was used to perform large-scale screening studies, exploring nearly 10000 unique chemistries via a DOE INCITE grant. from this data set, machine learning models were developed that can predict the coefficient of friction and adhesion force from only a SMILES representation of the film chemistry. Using the machine learning model, nearly 200,000 additional chemistries (taken from the ChEMBL small molecules database) were able to be explored in less than the time of just a single simulation.
Most recently, this prior work is being leveraged to design adaptable thin films that can dynamically switch their surface interactions to match the solvent. These systems could ultimately lead to breakthroughs in anti-fouling for anti-fouling coatings.
Related publications:
- Quach CD, Gilmer JB, Pert D, Mason-Hogans A, Iacovella CR, Cummings PT, McCabe C (2022) High-Throughput Screening of Tribological Properties of Monolayer Films using Molecular Dynamics and Machine Learning The Journal of Chemical Physics, 156, 15, 154902 DOI:10.1063/5.0080838
- Summers AZ, Gilmer JB, Iacovella CR, Cummings PT, McCabe C (2020) MoSDeF, a Python Framework Enabling Large-Scale Computational Screening of Soft Matter: Application to Chemistry-Propery Relationships in Lubricating Monolayer Films, Journal of Chemical Theory and Computation., 16,3,1779-1793 DOI:10.1021/acs.jctc.9b01183
- Black JE, Summers AZ, Iacovella CR, Cummings PT, McCabe CM (2019) Investigation of the Impact of Cross-Polymerization on the Structural and Frictional Properties of Alkylsilane Monolayers Using Molecular Simulation, Nanomaterials, 9(4), 639 DOI:10.3390/nano9040639
- Summers AZ, Iacovella CR, Cummings PT, McCabe C, (2017) Investigating Alkylsilane Monolayer Tribology at a Single-Asperity Contact with Molecular Dynamics Simulation, Langmuir, 33 (42), pp 11270–11280 DOI:10.1021/acs.langmuir.7b02479
- Summers AZ, Iacovella CR, Billingsley MR, Arnold ST, Cummings PT, McCabe C (2016) Influence of Surface Morphology on the Shear-Induced Wear of Alkylsilane Monolayers: Molecular Dynamics Study, Langmuir, 32(10), pp 2348-2359 DOI:10.1021/acs.langmuir.5b03862
- Klein C, Iacovella CR, McCabe C, Cummings PT (2015) Tunable Transition from Hydration to Monomer-Supported Lubrication in Zwitterionic Monolayers Revealed by Molecular Dynamics Simulation, Soft Matter, 11, pp 3340-3346 DOI:10.1039/C4SM02883J
- Black J, Iacovella CR, Cummings PT, McCabe C, (2015) Molecular Dynamics Study of Alkylsilane Monolayers on Realistic Amorphous Silica Surfaces, Langmuir, 31 (10), pp 3086–3093 DOI:10.1021/la5049858
Multiscale Modeling of the Stratum Corneum Layer of Skin
Skin acts as an important barrier between an organism and the outside environment, while also acting to help prevent dehydration. Links between skin disease and barrier function have been found, and there is now a growing consensus that this can have systemic impacts on overall health. The barrier function of skin is known to be localized within the outermost layer, the stratum corneum (SC). The SC is unique in that, unlike most biological membranes, it does not contain any phospholipids, instead being composed primarily of a mixture of ceramide lipids, free fatty acids, and cholesterol. This mixture of lipids is actually quite complex, with at least 14 unique ceramides found in mammalian skin, and a range of tail lengths for the free fatty acids.
To be able to model large system sizes, over long time scales to be able to allow membrane structures to arise spontaneously via self-assembly, coarse-grained (CG) models are required. A common approach to determining CG force fields is the iterative Boltzmann inversion (IBI) scheme, which relies upon self-consistently modifying the force field until the radial distribution function matches that of an equivalent atomistic target state. While effective, this scheme tends to produce force fields that only work at the state point and molecule for which they were derived, i.e., they are not transferrable. This is particularly an issue considering self-assembly is naturally a multistate process. To address this, an extension to the IBI approached, termed the multistate IBI (MS-IBI), was developed. In MS-IBI the CG force fields are iteratively updated to match multiple of target states simultaneously. These targets may be the same molecules under different thermodynamic conditions or different structures (e.g., bulk vs. bilayer configurations) or the same state but simulated using different thermodynamic ensembles (e.g., running NVT and NPT simultaneously enables the correct density pressure relationship to be achieved in the CG model). Targets may also be from different molecules but with the same CG building blocks, e.g., the tails in ceramides and free fatty acids contain the same chemical structure.
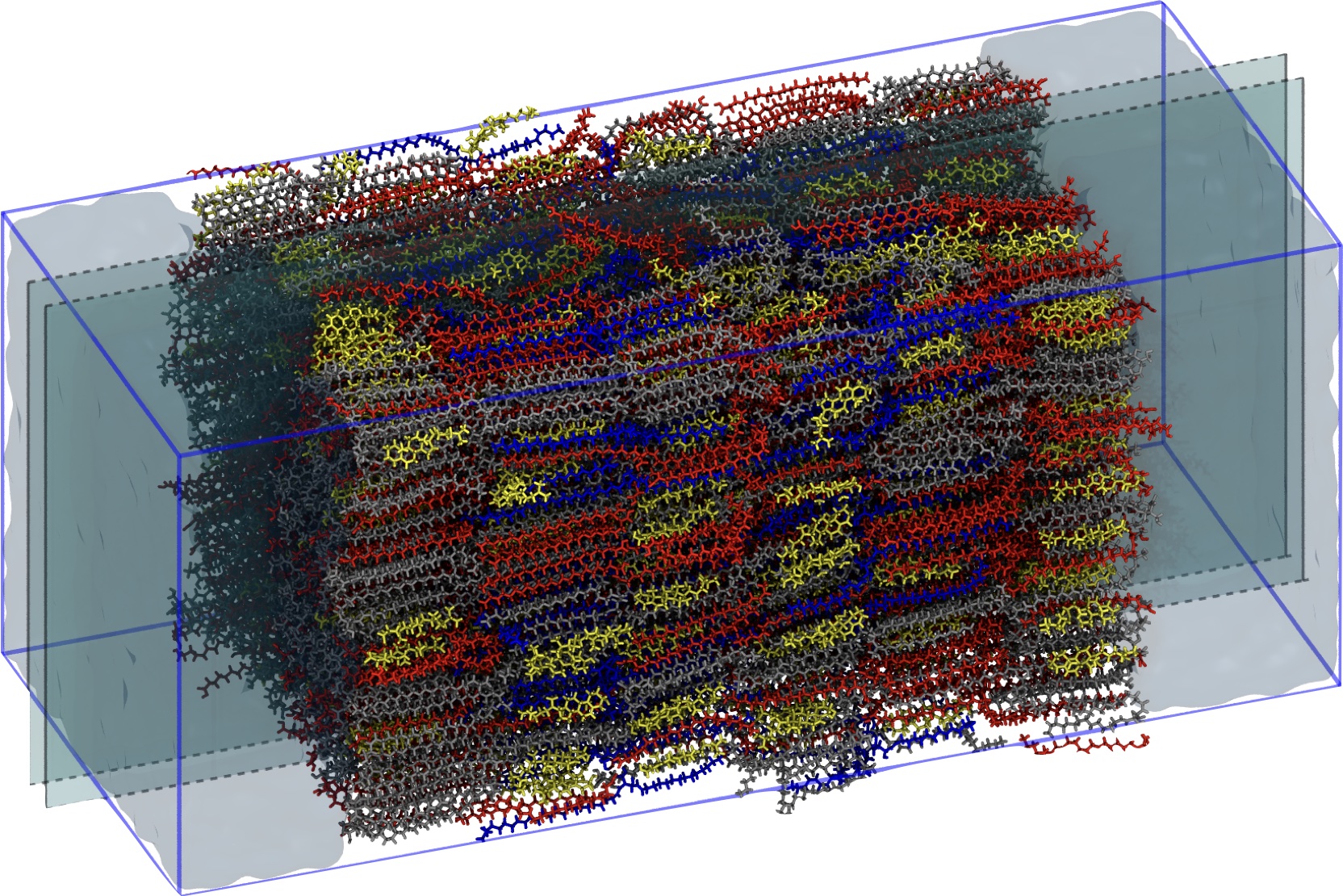
This approach enabled the development transferrable models for water, ceramide NS, free fatty acid, and cholesterol that are able to self-assemble multilayer structures. In fact, these CG force fields enabled the simulation ofthe first self-assembled model system of the short pperiodicty phase of the SC. The development of a reverse-mapping scheme to reinsert atomistic details, allows multiscale modeling of the SC, e.g., enabling examination of self-assembled multilayers but with atomistic level resolution (e.g., identifying hydrogen bonding and allowing comparison to experimental neutron scattering).
Most recently, the transferrability of the CG models developed via MS-IBI have been leveraged to allow for the modeling of a wide range of other ceramides to increase the complexity of our simulation models and provide additional examination of experimentally relevant systems.
Related publications:
- Shamaprasad P, Frame CO, Moore TC, Yang AH, Iacovella CR, Bouwstra JA, Bunge AL, McCabe C (2022) Using Molecular Simulation to Understand the Skin Barrier, Progress in Lipid Research, 88, 101184 DOI:10.1016/j.plipres.2022.101184
- Shamaprasad P, Moore TC, Via D, Iacovella CR, Bunge AL, McCabe C (2022) Multiscale Simulation of Ternary Stratum Corneum Lipid Mixtures: Effects of Cholesterol Composition, Langmuir 38, 24, 7496-7511 DOI:10.1021/acs.langmuir.2c00471
- Moore TC, Hartkamp R, Iacovella CR, Bunge AL, McCabe C (2018) The Influence of Ceramide Tail Length on the Structure of Bilayers Composed of Stratum Corneum Lipids, Biophysical Journal, 114(1) pp113-125 DOI:10.1016/j.bpj.2017.10.031
- Moore TC, Iacovella CR, Leonhard A, Bunge AL, McCabe C, (2017) Molecular Dynamics Simulations of Stratum Corneum Lipid Mixtures: A Multiscale Perspective, Biochemical and Biophysical Research Communications, 498, 2, 313-318 DOI:10.1016/j.bbrc.2017.09.040
- Moore TC, Iacovella CR, Hartkamp R, McCabe C, (2016) A Coarse-Grained Model of Stratum Corneum Lipids: Free Fatty Acids and Ceramide NS, The Journal of Physical Chemistry B, 120, pp 9944-9958 DOI:10.1021/acs.jpcb.6b08046
- Moore TC, Iacovella CR, McCabe C (2016) Development of a Coarse-grained Water Forcefield via Multistate Iterative Boltzmann Inversion, Molecular Modeling and Simulation: Applications and Perspectives, Foundations of Molecular Modeling and Simulation, pp 37-52 DOI:10.1007/978-981-10-1128-3_3
- Moore TC, Iacovella CR, McCabe C, (2014) Derivation of Coarse-grained Potentials via Multistate Iterative Boltzmann Inversion, The Journal of Chemical Physics, 140, 224104 DOI:10.1063/1.4880555
- Guo S, Moore TC, Iacovella CR, Strickland LA, McCabe C, (2013) Simulation Study of the Structure and Phase Behavior of Ceramide Bilayers and the Role of Lipid Head Group Chemistry, Journal of Chemical Theory and Computation, 9 (11), pp 5116–5126 DOI:10.1021/ct400431e